スケールフリー仮定への依存
すべての共発現ネットワークがスケールフリー構造を持つとは限りません。SGCRNAでは、この仮定に依存しない解析を目指します。
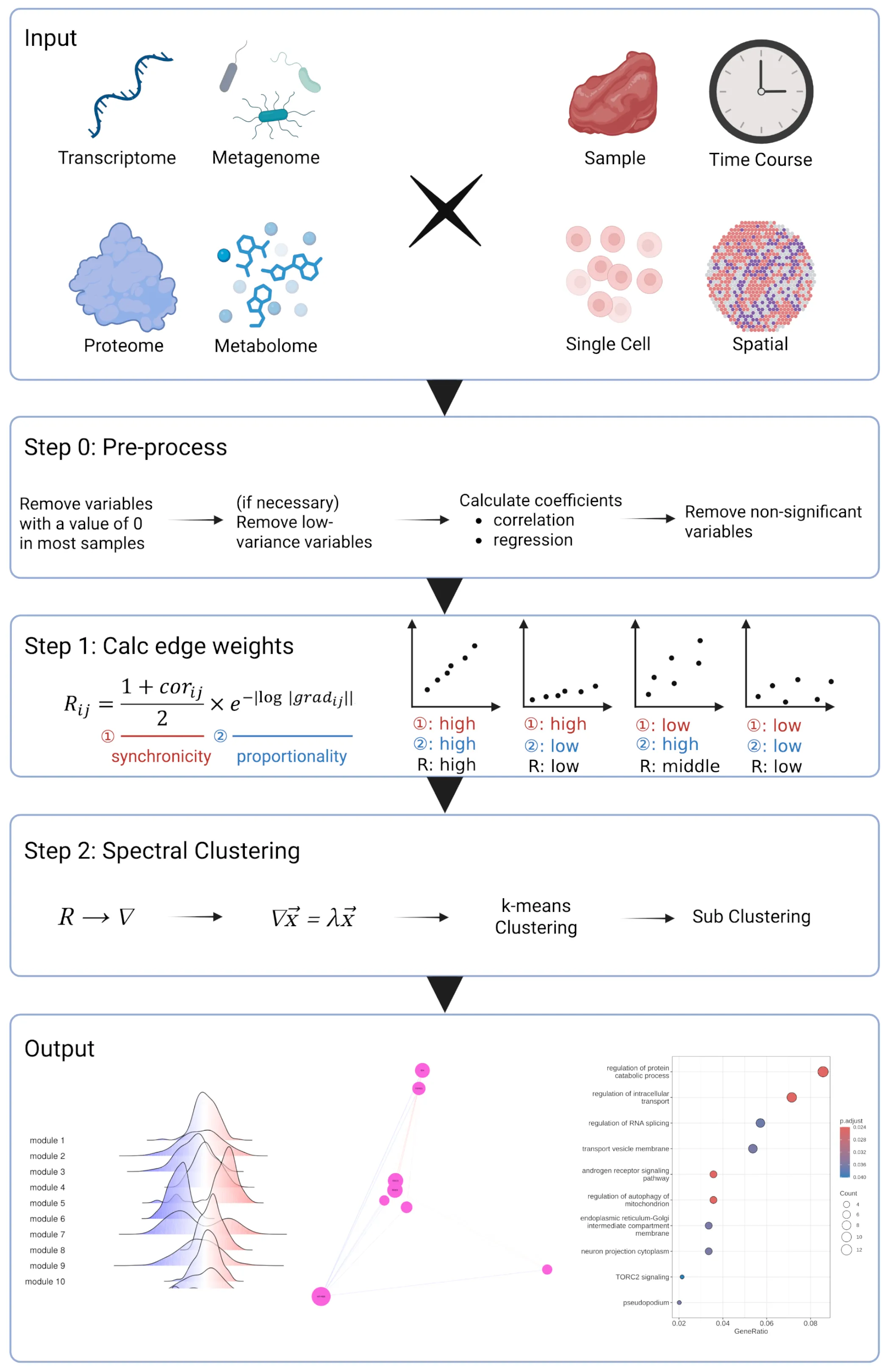
生命科学データの大規模化・多様化に対応するため、既存の解析手法を単に利用するだけでなく、 データの性質に合わせた新しいバイオインフォマティクス手法の開発にも取り組んでいます。 その一例として、共発現ネットワーク解析のための新手法 SGCRNA を開発しました。
SGCRNA は、遺伝子発現量などの多変量データから、互いに関連して変動する変数群をモジュールとして検出するための 共発現ネットワーク解析手法です。従来広く用いられてきた WGCNA では、ネットワークがスケールフリー性を持つという仮定、 ソフト閾値などのパラメータ調整、相関係数だけでは捉えにくい発現変動量の違いといった課題がありました。 SGCRNA は、相関係数に加えて回帰直線の傾きを考慮し、スペクトラルクラスタリングによってモジュールを検出することで、 これらの課題に対応することを目指しています。
共発現ネットワーク解析では、発現パターンが似ている遺伝子を同じモジュールとしてまとめることで、
生物学的に意味のある遺伝子群や候補バイオマーカーを探索します。
しかし、既存手法では解析対象のデータが想定されたネットワーク構造に合わない場合や、
パラメータの選択によって結果が変化する場合があります。
また、相関係数が高くても、一方の遺伝子の変動幅が極端に小さい場合には、生物学的に解釈しにくい関係を拾ってしまう可能性があります。
すべての共発現ネットワークがスケールフリー構造を持つとは限りません。SGCRNAでは、この仮定に依存しない解析を目指します。
従来法では、解析ごとにソフト閾値などを検討する必要があります。SGCRNAでは、より自動化されたモジュール検出を目指します。
高い相関があっても、変動幅が大きく異なる変数同士は、同じ制御単位として扱うべきか慎重に判断する必要があります。
transcriptome、spatial transcriptome、metagenomeなど、観測数や変数数が大きく異なるデータに対応できる設計が必要です。
SGCRNAでは、2つの変数の関係を評価する際に、相関係数だけでなく回帰係数の大きさも用います。 これにより、同じ方向に変動しているかどうかだけでなく、変動量の比がどの程度そろっているかを評価します。 その後、得られたスコア行列をラプラシアン行列に変換し、固有ベクトルに基づくスペクトラルクラスタリングを行います。
SGCRNAは、通常のbulk RNA-seqだけでなく、空間トランスクリプトーム、メタゲノム解析など、
さまざまな種類のデータに適用できるように設計しています。
研究では、公開データを用いて、モジュールの再現性、安定性、計算効率、生物学的妥当性を評価しました。

生命科学では、単一遺伝子の変化だけでなく、複数の遺伝子や分子がどのように協調して変動しているかを理解することが重要です。
SGCRNAのような新しいネットワーク解析手法を開発することで、従来法では見落とされていたモジュール、細胞種特異的な変動、
微生物間の相互作用などを、より解釈しやすい形で抽出できる可能性があります。
当分野では、このような手法開発を通じて、発生、再生、疾患などに関わる 複雑な生命現象をデータから理解することを目指しています。
本ページは、Briefings in Bioinformatics に掲載された SGCRNA に関する論文の内容に基づいています。 ソフトウェアおよび解析コードは公開リポジトリから利用できます。
SGCRNA: spectral clustering-guided co-expression network analysis without scale-free constraints for multi-omic data
Briefings in Bioinformatics, 2026
SGCRNAの解析に関連するコードはGitHubで公開されています。研究目的に応じて、再解析や手法の拡張に利用できます。